建立日期 44990
主題: 非典型溶血性尿毒症候群(aHUS)的準確診斷


Accurate Diagnosis in Patients With Thrombotic Microangiopathy/Atypical Hemolytic Uremic Syndrome (aHUS)

Dr. Arif Asif MD, Professor Arif Asif, MD, FASN, FNKF
Professor of Medicine and Chairman, Department of Medicine, Jersey Shore University Medical Center, Seton Hall-Hackensack-Meridian School of Medicine, New Jersey, USA
栓塞性微血管病變 (thrombotic microangiopathy, TMA) 診斷上有三大要素:血小板低下、溶血、以及標的器官損傷。非典型溶血性尿毒症候群 (atypical hemolytic uremic syndrome, aHUS) 是一種可發生在成人和兒童中,由基因突變導致補體失控、引起嚴重後果的疾病,屬於 TMA 的一種。由於近年不同類型的 TMA 已有不同治療策略,不應再統一以血漿置換進行處置,因此正確的鑑別診斷格外重要 1。
區分 aHUS 和其他類型的 TMA 非常重要,因為治療方案會有所不同
TMA 的 初 步 診 斷 首 先 仰 賴 微 血 管 溶 血 性 貧 血(Microangiopathic hemolytic anemia, MAHA) 與 血小板低下的確認 2-4,其中血小板低下不應只以固定界限值作為判斷依據 ( 如血小板數 < 150 x 109/ L),仍需考量患者基礎的血小板數量,若發現血小板數出現 >25 % 的降幅 ( 例如從 300 x 109/ L 降至 225 x 109/L),即使降低後的值仍高於界限值,仍應視為發生血小板低下 3。接下來的診斷要件是標的器官損傷,而其中務須注意 aHUS 的相關損傷並不只限於腎臟,腦部、心臟、肝臟、腸胃道、眼睛等任何器官都可能受到微血管栓塞的影響。無法只透過特定的器官損傷來區別各類型的 TMA,最近發展的 ADAMTS-13 酵素活性和志賀氏毒素檢測才是真正能鑑別 aHUS 與其他類 TMA 的關鍵檢查。若患者的 ADAMTS-13 酵素活性 > 5 % 且志賀氏毒素呈現陰性,則很有可能就患有 aHUS。
基因突變與補體增強事件 (complement amplifying conditions, CAC) 是 aHUS發病的危險因子
近年隨著基因遺傳學的快速發展,目前已在 aHUS 患者發現許多關鍵基因突變;這些突變多發生於補體調節蛋白(complement regulator proteins) 的基因上,不過目前仍有 40 - 50 % 的患者基因突變仍然不明,因此 aHUS現在還無法以基因突變進行篩檢和確診 5-9。此外,突變類型與預後似乎也不具相關性 6,10,而這方面的進展仍有限。值得注意的是,許多 aHUS 患者的家屬雖帶有相同的突變卻從未發病,顯示突變本身有時不足以驅動病理機制。目前認為突變帶原者大多在經歷補體增強事件(complement amplifying conditions, CAC) 強烈刺激補體系統後,基於患者根本上已有基因突變造成補體調節系統失控,而揭露了 aHUS 致病機制 11。近期的臨床研究主張約 69 % 的 aHUS 患者都曾經發生過 CAC6,像是惡性高血壓 (malignant hypertension, MHTN)、懷孕、自體免疫疾病、癌症、各種感染、手術、器官移植等等 13-16。
血漿置換不能解決 aHUS 患者潛在的慢性不受控制的補體活性
補體調節系統失控而造成補體過度活化是 aHUS 的主要致病原因 1,6,特別是補體替代路徑 (alternative pathway)的失調。補體替代路徑隨時保持低度活化,是人體面對病原體的先遣快打部隊,能迅速反應病原的侵襲 2,12。不過此補體替代路徑得有效地管控,來自肝臟產生的補體調節蛋 白 (complement regulator proteins) 扮演調控的角色,以維持體內補體的低度活化,而不會去攻擊自體的細胞 2,12。
當這些調節蛋白或其補體系統內所對應受體若發生基因突變而失能,補體系統經補體增強事件 (complement amplifying conditions, CAC) 活化後,無法控制的大量補體活化,而攻擊自體的血管內皮細胞,造成血管內皮細胞造成劇烈破壞 13-16,進而活化血小板並吸引凝血因子和纖維蛋白聚集,造成微小血管內血栓的生成。因此所有的器官就可能因為發生微小血管內血栓而導致組織缺氧及損傷 17,由此可知 aHUS 為什麼會造成各種器官傷害。
更重要的是,因 aHUS 的發病機制與其他類型 TMA 不同,故血漿置換在 aHUS 患者的預後不佳:臨床研究顯示 33– 40 % 的 aHUS 患者在第一次出現臨床症狀時就會直接進展至末期腎臟病 (ESRD) 或死亡 6,10,無論是否接受血漿置換也會有 65 % 的患者在診斷後一年內出現永久腎臟損傷、需進行透析或死亡 ( 圖一 )10,18,19 。
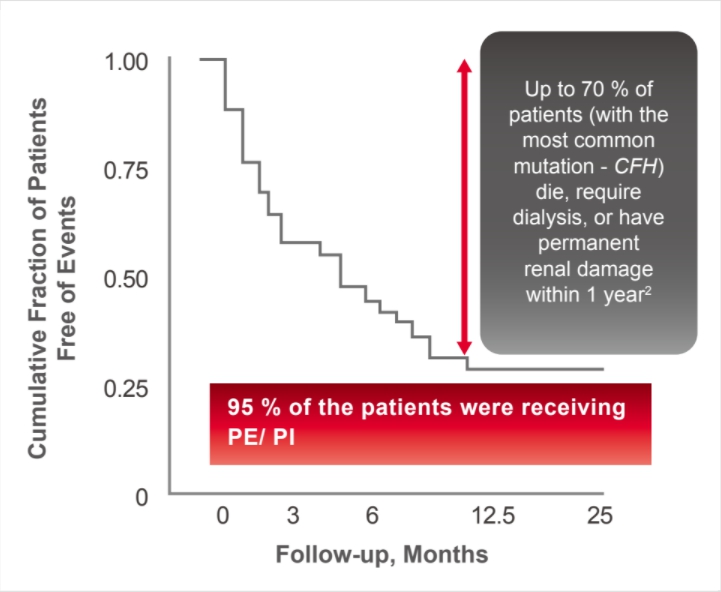 (圖一) 研究顯示在診斷後一年,儘管有將近 95 % 的患者接受血漿置換,但依然有將近 70 % 的 aHUS 患者死亡、發生永久腎臟損害或需進行透析。
(圖一) 研究顯示在診斷後一年,儘管有將近 95 % 的患者接受血漿置換,但依然有將近 70 % 的 aHUS 患者死亡、發生永久腎臟損害或需進行透析。從臨床試驗的結果可看出,血漿置換對於診斷一年後需透析的患者比率或ESRD 發生率 20、診斷後的 90 日死亡率 ( 圖二 )、住院死亡率、以及住院時間均無顯著效益 21。儘管血漿置換可能改善部分 aHUS 患者血中 LDH 的濃度和血小板低下等表面指標,但對於真正關鍵的標的器官受損率和死亡率卻完全沒有幫助;況且 aHUS 對腎臟的破壞程度明顯超越其他類型 TMA,若延誤治療往往會造成很嚴重的後果 22。
.jpg) (圖二) 研究顯示血漿置換對於診斷後的 90 日死亡率均無顯著效益。
(圖二) 研究顯示血漿置換對於診斷後的 90 日死亡率均無顯著效益。當補體增強事件 (CAC) 獲得控制,TMA 仍然持續,即應進行 aHUS 鑑別診斷
aHUS 其實是一種罕見疾病,因此在面對患者時,當下務必優先處置各項症狀和補體增強事件 (CAC);當補體增強事件 (CAC) 獲得控制,TMA 仍未獲改善,應依前述診斷原則利用 ADAMTS-13 酵素活性及志賀毒素檢測,來確診aHUS ( 圖三 )23。舉例來說,近期有一位懷孕三十三週的33 歲女性案例,因胎盤早剝大量出血也失去胎兒,同時伴有嚴重溶血 LDH 升高、血液抹片看到許多紅血球裂片、血小板低下、以及腎衰竭,ADAMTS-13 檢測結果為 56 %。在等待 ADAMTS-13 結果時先使用血漿治療,僅只有部分血液數值改善,但腎功能並沒有任何改變,確診為 aHUS 後就展開針對 aHUS 的單株抗體治療,治療兩周後患者血液數值及腎功能全部回復正常,也脫離洗腎。目前已持續治療了一年,患者的腎臟功能、血紅素值和血小板數仍維持正常 23。
另一例 43 歲的女性案例因視力受損和頭痛數天而求診,患者長期慢性頭痛及嚴重高血壓 ( 藥物無法控制 ),檢查結果顯示有嚴重溶血LDH 升高、血紅素數值低下、血液抹片看到許多紅血球裂片,但血小板數值正常,ADAMTS-13 檢測結果為64 %。等待 ADAMTS-13 結果時先使用血漿治療,確診為 aHUS 後就展開針對 aHUS 的單株抗體治療,隨即在各方面獲得改善 23。以上案例都是先針對其症狀和補體增強事件 (CAC) 試圖進行控制後,當控制效果不佳即思考 aHUS 的可能性,並執行鑑別診斷,確診為 aHUS後立刻使用單株抗體來治療。
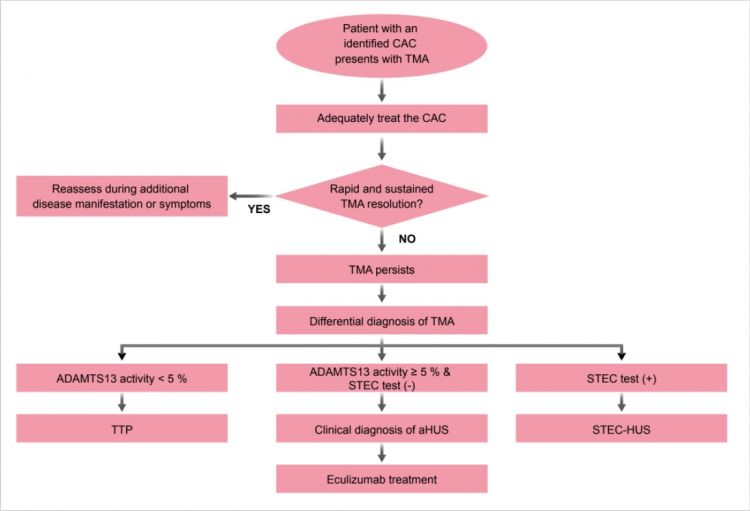
(圖三) aHUS 併有 CAC 患者的處置建議:先妥善治療 CAC 和症狀,當控制效果不佳應即思考 aHUS 的可能性,執行鑑別診斷 23。
會議筆記
• 法國研究 24 顯示近 60 % 與懷孕相關的 aHUS 會發生於第二次懷孕,所以對於有發病風險的患者,每次懷孕都必須特別注意,讓疾病惡化或標的器官損傷的可能性降到最低。
• 血漿置換可能改善部分 aHUS 患者的 LDH 或結合蛋白血中濃度或血小板數值等血液指標,因此有些醫師認為血漿置換對 aHUS 具有療效;但其實標的器官損傷與死亡率才是重點療效指標,而這兩項指標在有無使用血漿治療的患者並無明顯差異,顯示血漿治療無法真的改善 aHUS 病情和預後。
• aHUS 患者的高血壓主要是由腎臟血管所釋放的腎素 (renin) 所引起,當下很難進行控制。對於此類患者會先給予angiotensin-converting enzyme inhibitors (ACEI) / angiotensin receptor blockers (ARB) 藥物降低血壓,再針對 aHUS 的根本發病原因進行治療;因為高血壓也只是其中一種器官損傷的徵象,可能還有其他器官損傷會以不同型式表現,所以確診後必須盡速治本而不僅是治標。
• 不建議針對疑似患有 aHUS 的患者進行腎臟切片檢查,因 aHUS 患者發生出血或其他併發症的風險過高。若其他醫師已對患者做過腎臟切片檢查,其結果可作為參考,但不建議特地為患者進行切片檢查作為診斷 aHUS 的依據。
Reference
1. Laurence J et al. Atypical hemolytic uremic syndrome (aHUS): essential aspects of an accurate diagnosis. Clin Adv Hematol Oncol 2016; 14 Suppl 11:2-15.
2. Laurence J. Atypical hemolytic uremic syndrome (aHUS): making the diagnosis. Clin Adv Hematol Oncol 2012;10:1-12.
3. Noris M & Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med 2009;361:1676-87.
4. Noris M et al. Complement factor H mutation in familial thrombotic thrombocytopenic purpura with ADAMTS13 deficiency and renal involvement. J Am Soc Nephrol 2005;16:1177-83.
5. Bresin E et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype.J Am Soc Nephrol 2013; 24:475-86.
6. Noris M et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 2010;5:1844-59.
7. Fremeaux-Bacchi V et al. Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol 2013; 8:554-62.
8. Lemaire M et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 2013; 45:531-6.
9. Sanchez Chinchilla D et al. Complement mutations in diacylglycerol kinase-ε-associated atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 2014; 9:1611-9.
10. Caprioli J et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 20 06; 108:1267-79.
11. Kavanagh D et al. Atypical haemolytic uraemic syndrome. Br Med Bull 2006 ; 77-78:5-22.
12. Noris M et al. STEC-HUS, atypical HUS and TTP are all diseases of complement activation. Nat Rev Nephrol 2012; 8:622-33.
13. Nester CM et al. Atypical aHUS: State of the art. Mol Immunol 2015; 67 :31-42.
14. Riedl M et al. Spectrum of complement-mediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost 2014; 40:444-64.
15. Tsai HM. A mechanistic approach to the diagnosis and management of atypical hemolytic uremic syndrome. Transfus Med Rev 2014; 28:187-97.
16. Sperati CJ & Moliterno AR. Thrombotic microangiopathy: focus on atypical hemolytic uremic syndrome. Hematol Oncol Clin North Am 2015; 29:541-59.
17. Nayer A & Asif A. Atypical hemolytic-uremic syndrome: the interplay between complements and the coagulation system. Iran J Kidney Dis 2013; 7:340-5.
18. Sallée M et al. Myocardial infarction is a complication of factor H-associated atypical HUS. Nephrol Dial Transplant 2010; 25:2028-32.
19. Noris M et al. Atypical hemolytic-uremic syndrome. N Engl J Med 200 9; 361:1676-87.
20. Fakhouri F et al. Insights from the use in clinical practice of eculizumab in adult patients with atypical hemolytic uremic syndrome affecting the native kidneys: an analysis of 19 cases. Am J Kidney Dis 2 014; 63:40-8.
21. Li A et al. Treatment with or without plasma exchange for patients with acquired thrombotic microangiopathy not associated with severe ADAMTS13 deficiency: a propensity score-matched study. Transfusion 2016; 56:2069-77.
22. Pishko AM. Atypical Hemolytic Uremic Syndrome (aHUS): Essential Aspects of an Accurate Diagnosis. Blood 2014; 124:Abstract 4192.
23. Asif A et al. Atypical hemolytic uremic syndrome in the setting of complement-amplifying conditions: case reports and a review of the evidence for treatment with eculizumab. J Nephrol 2017; 30:347-62.
24. Fakhouri F et al. Pregnancy-associated hemolytic uremic syndrome revisited in the era of complement gene mutation. J Am Soc Nephrol 2010; 21:859-67.